Estuve buscando un programa que realice gráficos mostrando los niveles de transiciones y tranversiones en secuencias de ADN.
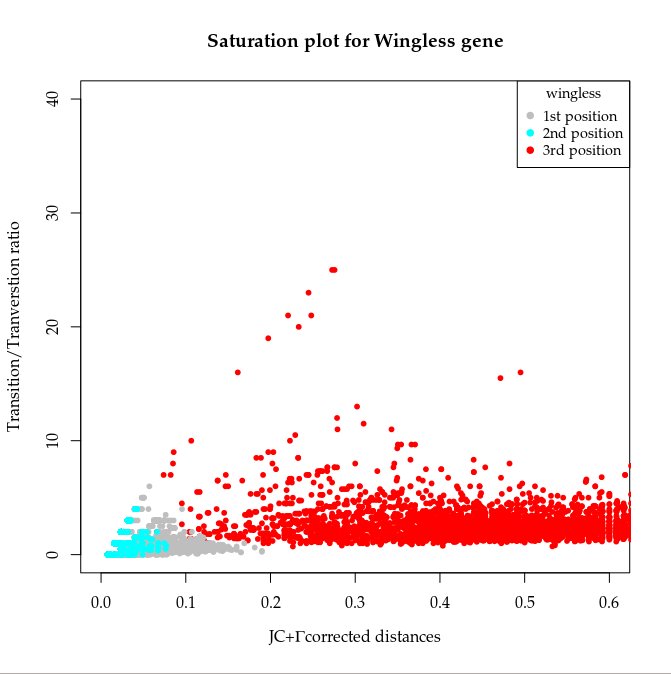
Se supone que el tercer codon de una secuencia de ADN tiende a llegar a "saturación" debido a que el código genético es degenerado (varios tripletes codifican el mismo aminoácido). Esta saturación ocurre cuando las mutaciones en el tercer codón han sido tan frecuentes que ya no llevan información filogenética. Es decir, se llega al grado en que dos secuencias saturadas son parecidas simplemente por chance.
El programa
DAMBE realiza este tipo de gráficos al plotear la relación de transiciones y transversiones versus distancia genética, pero no pude hacer funcionar la versión para Linux.
Encontré una función escrita para el
programa estadístico R que sí me funcionó.
La versión original la pueden encontrar aquí:
http://the-praise-of-insects.blogspot.fi/2010/04/transitions-in-r-redux.html
Download sample data here: wg_sample_data.zip
library(ape)
# modified from
# http://the-praise-of-insects.blogspot.fi/2010/04/transitions-in-r-redux.html
par(mfcol=c(1,1)); par(mar=c())
par(xpd=F, mar=c(5,4,4,2)+0.2, family="Palatino")
#Input: dat---an object of class 'DNAbin'
titv<-function(dat){
mat<-as.matrix(dat)
res<-matrix(NA, ncol=dim(mat)[1], nrow=dim(mat)[1], dimnames=list(x=names(dat), y=names(dat)))
for(i in 1:(dim(mat)[1] - 1)){
for(j in (i+1):dim(mat)[1]){
vec<-as.numeric(mat[i,])+as.numeric(mat[j,])-8
res[j,i]<-sum(!is.na(match(vec,c(200,56))))#Transitions
res[i,j]<-sum(!is.na(match(vec,c(152,168,88,104))))#Transversions
}
}
res
}
create_plots<-function(files) {
for( i in 1:length(files)) {
codon <- as.DNAbin(read.nexus.data(files[i]))
ti<-titv(codon)
tv<-t(ti)
if (i == 1) {
R1 <- ti[lower.tri(ti)]/tv[lower.tri(tv)]
dist1 <- dist.dna(codon, model="JC69", gamma=T, pairwise.deletion=T)
}
else if( i == 2) {
R2 <- ti[lower.tri(ti)]/tv[lower.tri(tv)]
dist2 <- dist.dna(codon, model="JC69", gamma=T, pairwise.deletion=T)
}
else if( i == 3) {
R3 <- ti[lower.tri(ti)]/tv[lower.tri(tv)]
dist3 <- dist.dna(codon, model="JC69", gamma=T, pairwise.deletion=T)
}
else {
print("error")
}
}
# print plot
xname = expression("JC+" * Gamma * "corrected distances")
yname = "Transition/Tranverstion ratio"
plot(NA, xlim=c(0,.6), ylim=c(0,40), xlab=xname, ylab=yname, main="Saturation plot for Wingless gene")
points(R3~dist3, xlim=c(0,.6), ylim=c(0,40), col="red", pch=20, xlab="", ylab="")
points(R1~dist1, xlim=c(0,.6), ylim=c(0,40), col="grey", pch=20, xlab="", ylab="")
points(R2~dist2, xlim=c(0,.6), ylim=c(0,40), col="cyan", pch=20, xlab="", ylab="")
legend.txt <- c("1st position", "2nd position", "3rd position")
legend.colors <- c("grey", "cyan", "red")
legend("topright", legend.txt, pch=19, col=legend.colors, title="wingless", cex=0.9)
}
files <- c("wg_1.nex", "wg_2.nex", "wg_3.nex");
create_plots(files);
Y este es el resultado final para el gen
wingless:
 |
Es importante el orden al plotear los puntos (primero los más abundantes).
Así es posible ver todos. |